ipyrad command line tutorial - Part I
This is the first part of the full tutorial for the command line interface (CLI) for ipyrad. In this tutorial we’ll walk through the entire assembly and analysis process. This is meant as a broad introduction to familiarize users with the general workflow, and some of the parameters and terminology. We will use as an example in this tutorial the Anolis data set from the first part of class. However, you can follow along with one of the other example data sets if you like and although your results will vary the procedure will be identical.
If you are new to RADseq analyses, this tutorial will provide a simple overview of how to execute ipyrad, what the data files look like, how to check that your analysis is working, and what the final output formats will be. We will also cover how to run ipyrad on a cluster and to do so efficiently.
Each grey cell in this tutorial indicates a command line interaction.
Lines starting with $ indicate a command that should be executed
in a terminal connected to the Habanero cluster, for example by copying and
pasting the text into your terminal. Elements in code cells surrounded
by angle brackets (e.g.
## Example Code Cell.
## Create an empty file in my home directory called `watdo.txt`
$ touch ~/watdo.txt
## Print "wat" to the screen
$ echo "wat"
wat
Overview of Assembly Steps
Very roughly speaking, ipyrad exists to transform raw data coming off the sequencing instrument into output files that you can use for downstream analysis.
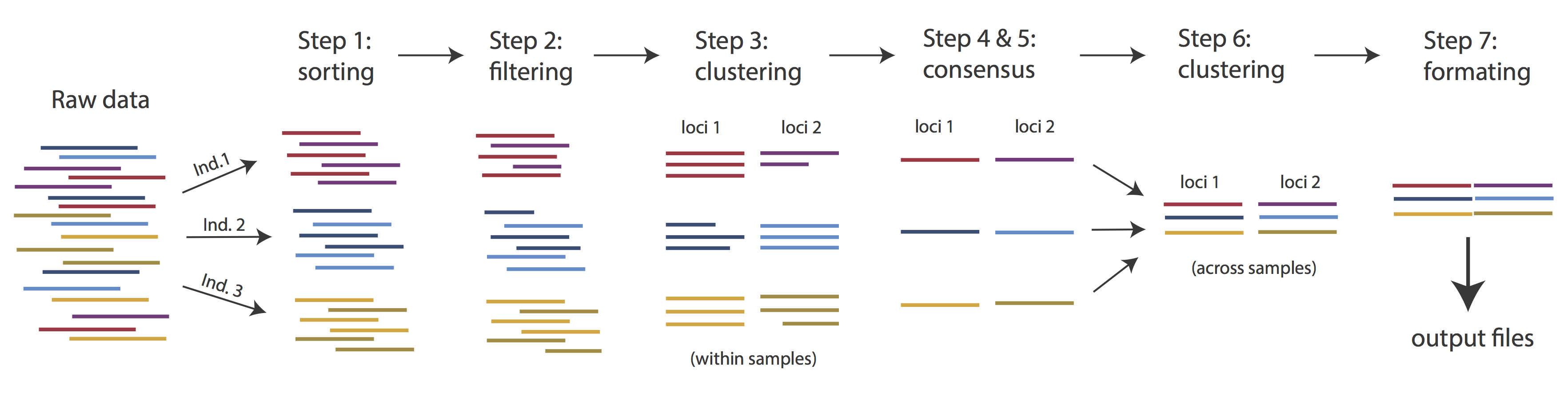
The basic steps of this process are as follows:
- Step 1 - Demultiplex/Load Raw Data
- Step 2 - Trim and Quality Control
- Step 3 - Cluster or reference-map within Samples
- Step 4 - Calculate Error Rate and Heterozygosity
- Step 5 - Call consensus sequences/alleles
- Step 6 - Cluster across Samples
- Step 7 - Apply filters and write output formats
Note on files in the project directory: Assembling rad-seq type sequence data requires a lot of different steps, and these steps generate a lot of intermediary files. ipyrad organizes these files into directories, and it prepends the name of your assembly to each directory with data that belongs to it. One result of this is that you can have multiple assemblies of the same raw data with different parameter settings and you don’t have to manage all the files yourself! (See Branching assemblies for more info). Another result is that you should not rename or move any of the directories inside your project directory, unless you know what you’re doing or you don’t mind if your assembly breaks.
Getting Started
If you haven’t already installed ipyrad go here first: installation
Working with the cluster
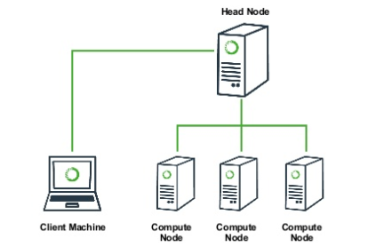
A typical high performance computing (HPC) cluster architecture looks somewhat like this, just with many many more “compute nodes”. The “head node” (or login node) is normally the only system that you will interact with. If you want to run a big job on the cluster, you will connect to the head node (with ssh), and will submit a job to the ‘work queue’. In this job you specify how many cores you want to run on, how much RAM you want, and how much time you think it’ll take. Then the work queue looks at your job and at all the other jobs in the queue and figures out when is the fairest time to start your job running. This can be almost immediately, or your job might sit in the queue for hours or days, if the system is very busy. Either way, you will rarely actually “see” your job run because you normally aren’t given access to the compute nodes directly.
This is usually okay, because usually if you want to run a “big” job, this means you want to run tons of small, quick tasks, or one or a few really really huges and slow tasks, and you kind of don’t care what’s happening, so long as they finish at some point. For us, for the benefit of exposing and monitoring the processes for the tutorial, having jobs locked away in the work queue is inconvenient. Fortunately, many HPC systems provide an “interactive” mode, which allows you to run certain limited tasks inside a terminal directly on one of the compute nodes.
Therefore, we will run most of this tutorial on assembly and analysis on the Habanero cluster
inside an “interactive” job. This will allow us to run our proccesses on
compute nodes, but still be able to remain at the command line so
we can easily monitor the progress. If you do not still have an active
ssh window on the cluster, begin by re-establishing the connection
through puTTY (Windows) or
ssh (Mac/Linux):
$ ssh <username>@habanero.rcs.columbia.edu
Submitting an interactive job to the cluster
Now we will submit an interactive job with relatively modest resource requests. Every cluster has different limits on its resources in terms of what is available to you and for how long. We could find these limits for the Columbia Habanero cluster by googling it. In this case we will each request that 4 cores be made available to us for 1 hour. Because each node has 24 cores this means that multiple people will actually be sharing the same node, which is not a problem.
# --pty tells it to connect us to compute nodes interactively
# --account tells it which account's resources to use
# --reservation tells it to use the resources on edu reserved for us
# -t tells it how much time to connect for
# /bin/bash tells it to open a bash terminal when we connect.
$ srun --pty --account=edu --reservation=edu_23 -t 1:00:00 -c 4 /bin/bash
Depending on cluster usage the job submission script can take more or less time to start. Because we have these resources reserved for us there should be very little wait time. Once your job starts your terminal will show that you are now connected to a compute node.
Inspecting running cluster processes
At any time you can ask the cluster for the status of your jobs with the
squeue command. This will list every running job on the cluster which can be
a pain to sort through. So for efficiency you can add the argument
-u <username> to limit it to showing just your jobs.
## Check the status of my running job on the 'proto' Queue
$ squeue -u work1
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
8367625 edu2 bash work1 R 0:09 1 node215
This confirms that the job we submitted started, that it’s running on node215, etc.
ipyrad help
To better understand how to use ipyrad, let’s take a look at the help argument. We will use some of the ipyrad arguments in this tutorial (for example: -n, -p, -s, -c, -r). But, the complete list of optional arguments and their explanation is below.
$ ipyrad --help
usage: ipyrad [-h] [-v] [-r] [-f] [-q] [-d] [-n new] [-p params]
[-b [branch [branch ...]]] [-m [merge [merge ...]]] [-s steps]
[-c cores] [-t threading] [--MPI] [--preview]
[--ipcluster [ipcluster]] [--download [download [download ...]]]
optional arguments:
-h, --help show this help message and exit
-v, --version show program's version number and exit
-r, --results show results summary for Assembly in params.txt and
exit
-f, --force force overwrite of existing data
-q, --quiet do not print to stderror or stdout.
-d, --debug print lots more info to ipyrad_log.txt.
-n new create new file 'params-{new}.txt' in current
directory
-p params path to params file for Assembly:
params-{assembly_name}.txt
-b [branch [branch ...]]
create a new branch of the Assembly as
params-{branch}.txt
-m [merge [merge ...]]
merge all assemblies provided into a new assembly
-s steps Set of assembly steps to perform, e.g., -s 123
(Default=None)
-c cores number of CPU cores to use (Default=0=All)
-t threading tune threading of binaries (Default=2)
--MPI connect to parallel CPUs across multiple nodes
--preview run ipyrad in preview mode. Subset the input file so
it'll runquickly so you can verify everything is
working
--ipcluster [ipcluster]
connect to ipcluster profile (default: 'default')
--download [download [download ...]]
download fastq files by accession (e.g., SRP or SRR)
* Example command-line usage:
ipyrad -n data ## create new file called params-data.txt
ipyrad -p params-data.txt ## run ipyrad with settings in params file
ipyrad -p params-data.txt -s 123 ## run only steps 1-3 of assembly.
ipyrad -p params-data.txt -s 3 -f ## run step 3, overwrite existing data.
* HPC parallelization across 32 cores
ipyrad -p params-data.txt -s 3 -c 32 --MPI
* Print results summary
ipyrad -p params-data.txt -r
* Branch/Merging Assemblies
ipyrad -p params-data.txt -b newdata
ipyrad -m newdata params-1.txt params-2.txt [params-3.txt, ...]
* Subsample taxa during branching
ipyrad -p params-data.txt -b newdata taxaKeepList.txt
* Download sequence data from SRA into directory 'sra-fastqs/'
ipyrad --download SRP021469 sra-fastqs/
* Documentation: http://ipyrad.readthedocs.io
Create a new parameters file
ipyrad uses a text file to hold all the parameters for a given assembly.
Start by creating a new parameters file with the -n flag. This flag
requires you to pass in a name for your assembly. In the example we use
anolis but the name can be anything at all. Once you start
analysing your own data you might call your parameters file something
more informative, like the name of your organism and some details on the settings.
# go to our working directory
$ cd ~/ipyrad-workshop
# create a new params file named 'anolis' (Or the name of an alternative library)
$ ipyrad -n anolis
This will create a file in the current directory called params-anolis.txt. The params file lists on each line one parameter followed by a ## mark, then the name of the parameter, and
then a short description of its purpose. Lets take a look at it.
$ cat params-anolis.txt
------- ipyrad params file (v.0.7.28)-------------------------------------------
anolis ## [0] [assembly_name]: Assembly name. Used to name output directories for assembly steps
./ ## [1] [project_dir]: Project dir (made in curdir if not present)
## [2] [raw_fastq_path]: Location of raw non-demultiplexed fastq files
## [3] [barcodes_path]: Location of barcodes file
## [4] [sorted_fastq_path]: Location of demultiplexed/sorted fastq files
denovo ## [5] [assembly_method]: Assembly method (denovo, reference, denovo+reference, denovo-reference)
## [6] [reference_sequence]: Location of reference sequence file
rad ## [7] [datatype]: Datatype (see docs): rad, gbs, ddrad, etc.
TGCAG, ## [8] [restriction_overhang]: Restriction overhang (cut1,) or (cut1, cut2)
5 ## [9] [max_low_qual_bases]: Max low quality base calls (Q<20) in a read
33 ## [10] [phred_Qscore_offset]: phred Q score offset (33 is default and very standard)
6 ## [11] [mindepth_statistical]: Min depth for statistical base calling
6 ## [12] [mindepth_majrule]: Min depth for majority-rule base calling
10000 ## [13] [maxdepth]: Max cluster depth within samples
0.85 ## [14] [clust_threshold]: Clustering threshold for de novo assembly
0 ## [15] [max_barcode_mismatch]: Max number of allowable mismatches in barcodes
0 ## [16] [filter_adapters]: Filter for adapters/primers (1 or 2=stricter)
35 ## [17] [filter_min_trim_len]: Min length of reads after adapter trim
2 ## [18] [max_alleles_consens]: Max alleles per site in consensus sequences
5, 5 ## [19] [max_Ns_consens]: Max N's (uncalled bases) in consensus (R1, R2)
8, 8 ## [20] [max_Hs_consens]: Max Hs (heterozygotes) in consensus (R1, R2)
4 ## [21] [min_samples_locus]: Min # samples per locus for output
20, 20 ## [22] [max_SNPs_locus]: Max # SNPs per locus (R1, R2)
8, 8 ## [23] [max_Indels_locus]: Max # of indels per locus (R1, R2)
0.5 ## [24] [max_shared_Hs_locus]: Max # heterozygous sites per locus (R1, R2)
0, 0, 0, 0 ## [25] [trim_reads]: Trim raw read edges (R1>, <R1, R2>, <R2) (see docs)
0, 0, 0, 0 ## [26] [trim_loci]: Trim locus edges (see docs) (R1>, <R1, R2>, <R2)
p, s, v ## [27] [output_formats]: Output formats (see docs)
## [28] [pop_assign_file]: Path to population assignment file
In general the defaults are sensible, and we won’t mess with them for now, but there are a few parameters we must change: the path to the raw data, the dataype, and the restriction overhang sequence.
We will use the nano text editor to modify params-anolis.txt and change
these parameters:
$ nano params-anolis.txt
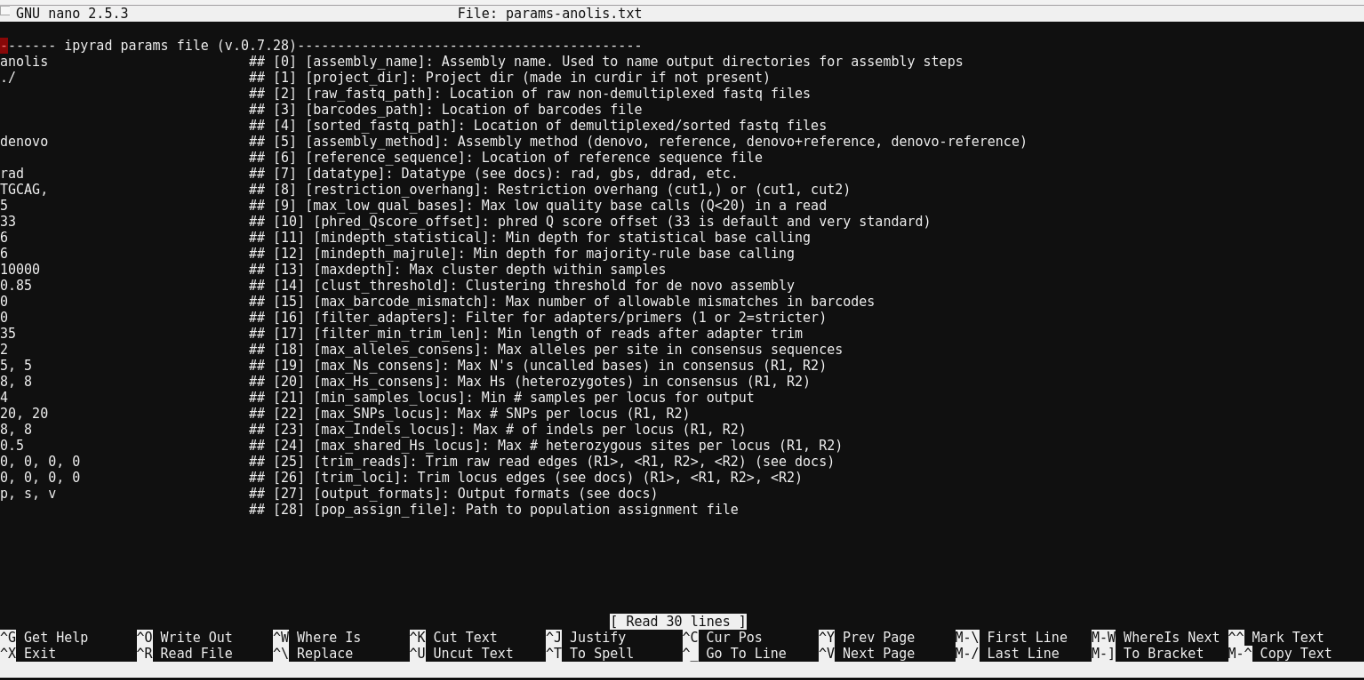
Nano is a command line editor, so you’ll need to use only the arrow keys on the keyboard for navigating around the file. Nano accepts a few special keyboard commands for doing things other than modifying text, and it lists these on the bottom of the frame.
We need to specify where the raw data files are located, the type of data we are using (.e.g., ‘gbs’, ‘rad’, ‘ddrad’, ‘pairddrad), and which enzyme cut site overhangs are expected to be present on the reads. We list the parameter settings for three different empirical libraries below. Choose just one for your example analysis.
# Anolis data set
anoles ## [2] project_dir
/rigel/edu/radcamp/files/anoles/*.gz ## [4] [sorted_fastq_path]: Location of demultiplexed/sorted fastq files
gbs ## [7] [datatype]: Datatype (see docs): rad, gbs, ddrad, etc.
TGCAT, ## [8] [restriction_overhang]: Restriction overhang (cut1,) or (cut1, cut2)
# Pedicularis data set
pedicularis ## [2] project_dir
/rigel/edu/radcamp/files/SRP021469/*.gz ## [4] [sorted_fastq_path]: Location of demultiplexed/sorted fastq files
rad ## [7] [datatype]: Datatype (see docs): rad, gbs, ddrad, etc.
TGCAG, ## [8] [restriction_overhang]: Restriction overhang (cut1,) or (cut1, cut2)
# Finch data set
finch ## [2] project_dir
/rigel/edu/radcamp/files/SRP059199/*.gz ## [4] [sorted_fastq_path]: Location of demultiplexed/sorted fastq files
ddrad ## [7] [datatype]: Datatype (see docs): rad, gbs, ddrad, etc.
CCTGCAGG,AATTC ## [8] [restriction_overhang]: Restriction overhang (cut1,) or (cut1, cut2)
After you change these parameters you may save and exit nano by typing CTRL+o (to write Output), and then CTRL+x (to eXit the program).
Note: The
CTRL+xnotation indicates that you should hold down the control key (which is often styled ‘ctrl’ on the keyboard) and then push ‘x’.
Once we start running the analysis ipyrad will create several new
directories to hold the output of each step for this assembly. By
default the new directories are created in the project_dir
directory and use the prefix specified by the assembly_name parameter.
Because we use ./ for the project_dir for this tutorial, all these
intermediate directories will be of the form: ~/ipyrad-workshop/anolis_*,
or the analagous name that you used for your assembly name.
Note: Again, the
./notation indicates the current working directory. You can always view the current working directory with thepwdcommand (print working directory).
Input data format
Before we get started let’s take a look at what the raw data looks like.
Your input data will be in fastQ format, usually ending in .fq,
.fastq, .fq.gz, or .fastq.gz. The file/s may be compressed with
gzip so that they have a .gz ending, but they do not need to be. When loading
pre-demultiplexed data (as we are with the Anolis data) the location
of raw sample files should be entered on line 3 of the params file. Below are the
first three reads of one of the Anolis files.
## For your personal edification here is what this is doing:
## gunzip -c: Tells gzip to unzip the file and write the contents to the screen
## head -n 12: Grabs the first 12 lines of the fastq file. Fastq files
## have 4 lines per read, so the value of `-n` should be a multiple of 4
$ zcat /rigel/edu/radcamp/files/SRP021469/29154_superba_SRR1754715.fastq.gz | head -n 20
@D00656:123:C6P86ANXX:8:2201:3857:34366 1:Y:0:8
TGCATGTTTATTGTCTATGTAAAAGGAAAAGCCATGCTATCAGAGATTGGCCTGGGGGGGGGGGGCAAATACATGAAAAAGGGAAAGGCAAAATG
+
;=11>111>1;EDGB1;=DG1=>1:EGG1>:>11?CE1<>1<1<E1>ED1111:00CC..86DG>....//8CDD/8C/....68..6.:8....
@D00656:123:C6P86ANXX:8:2201:5076:34300 1:N:0:8
TGCATATGAACCCCAACCTCCCCATCACATTCCACCATAGCAATCAGTTTCCTCTCTTCCTTCTTCTTGACCTCTCCACCTCAAAGGCAACTGCA
+
@;BFGEBCC11=/;/E/CFGGGG1ECCE:EFDFCGGGGGGG11EFGGGGGCGG:B0=F0=FF0=F:FG:FDG00:;@DGGDG@0:E0=C>DGCF0
@D00656:123:C6P86ANXX:8:2201:5042:34398 1:N:0:8
TGCATTCAAAGGGAGAAGAGTACAGAAACCAAGCACATATTTGAAAAATGCAAGATCGGAAGAGCGGTTCAGCAGGAATGCCGAGACCGATCTCG
+
GGGGGGGCGGGGGGGGGGGGGEGGGFGGGGGGEGGGGGGGGGGGGGFGGGEGGGGGGGGGGGGGGGGGGGGGGGGGGGEGGGGGGGGG@@DGGGG
Each read is composed of four lines. The first is the name of the read (its location on the plate). The second line contains the sequence data. The third line is unused. And the fourth line is the quality scores for the base calls. The FASTQ wikipedia page has a good figure depicting the logic behind how quality scores are encoded.
The Anolis data are 96bp single-end reads prepared as GBS. The first five bases (TGCAT) form the restriction site overhang. All following bases make up the sequence data.
Step 1: Loading the raw data files
With reads already demultiplexed to samples, step 1 simply scans through the raw data, verifies the input format, and counts reads per sample. It doesn’t create any new directories or modify the raw files in any way.
Note on step 1: More commonly, rather than returning demultiplexed samples as we have here, sequencing facilities will give you one giant .gz file that contains all the sequences from your run. This situation only slightly modifies step 1, and does not modify further steps, so we will refer you to the full ipyrad tutorial for guidance in this case.
Now lets run step 1! For the Anolis data this will take <1 minute.
Special Note: In interactive mode please be aware to always specify the number of cores with the
-cflag. If you do not specify the number of cores ipyrad assumes you want all of them, but in this case you only have as many cores available as we requested when we started the interactive session. This can cause some confusion that will slow things down a bit. So specify the number of cores that you know are available in this case when using interactive mode.
## -p the params file we wish to use
## -s the step to run
## -c the number of cores to allocate <-- Important!
$ ipyrad -p params-anolis.txt -s 1 -c 4
-------------------------------------------------------------
ipyrad [v.0.7.28]
Interactive assembly and analysis of RAD-seq data
-------------------------------------------------------------
New Assembly: anolis
establishing parallel connection:
host compute node: [4 cores] on darwin
Step 1: Loading sorted fastq data to Samples
[####################] 100% loading reads | 0:00:04
10 fastq files loaded to 10 Samples.
In-depth operations of running an ipyrad step
Any time ipyrad is invoked it performs a few housekeeping operations:
- Load the assembly object - Since this is our first time running any steps we need to initialize our assembly.
- Start the parallel cluster - ipyrad uses a parallelization library called ipyparallel. Every time we start a step we fire up the parallel clients. This makes your assemblies go smokin’ fast.
- Do the work - Actually perform the work of the requested step(s) (in this case loading in sample reads).
- Save, clean up, and exit - Save the state of the assembly, and spin down the ipyparallel cluster.
As a convenience ipyrad internally tracks the state of all your steps in your
current assembly, so at any time you can ask for results by invoking the -r flag.
We also use the -p arg to tell is which params file (i.e., which assembly) we
want it to print stats for.
## -r fetches informative results from currently executed steps
$ ipyrad -p params-anolis.txt -r
Summary stats of Assembly anolis
------------------------------------------------
state reads_raw
punc_IBSPCRIB0361 1 250000
punc_ICST764 1 250000
punc_JFT773 1 250000
punc_MTR05978 1 250000
punc_MTR17744 1 250000
punc_MTR21545 1 250000
punc_MTR34414 1 250000
punc_MTRX1468 1 250000
punc_MTRX1478 1 250000
punc_MUFAL9635 1 250000
Full stats files
------------------------------------------------
step 1: ./anolis_s1_demultiplex_stats.txt
step 2: None
step 3: None
step 4: None
step 5: None
step 6: None
step 7: None
If you want to get even more info ipyrad tracks all kinds of wacky stats and saves them to a file inside the directories it creates for each step. For instance to see full stats for step 1 (the wackyness of the step 1 stats at this point isn’t very interesting, but we’ll see stats for later steps are more verbose):
$ cat anolis_s1_demultiplex_stats.txt
reads_raw
punc_IBSPCRIB0361 250000
punc_ICST764 250000
punc_JFT773 250000
punc_MTR05978 250000
punc_MTR17744 250000
punc_MTR21545 250000
punc_MTR34414 250000
punc_MTRX1468 250000
punc_MTRX1478 250000
punc_MUFAL9635 250000
Step 2: Filter reads
This step filters reads based on quality scores and maximum number of uncalled bases, and can be used to detect Illumina adapters in your reads, which is sometimes a problem under couple different library prep scenarios. Recalling from our exploration of the data with FastQC we have some problem with adapters, and a little noise toward the 3’ end. To account for this we will trim reads to 75bp and set adapter filtering to be quite aggressive.
Note: Trimming to 75bp seems a bit aggressive too, and based on the FastQC results you probably would not want to do this with if these were your real data. However, it will speed up the analysis considerably. Here, we are just trimming the reads for the sake of this workshop.
Edit your params file again with nano:
nano params-anolis.txt
and change the following two parameter settings:
2 ## [16] [filter_adapters]: Filter for adapters/primers (1 or 2=stricter)
0, 75, 0, 0 ## [25] [trim_reads]: Trim raw read edges (R1>, <R1, R2>, <R2) (see docs)
Note: Saving and quitting from
nano:CTRL+othenCTRL+x
$ ipyrad -p params-anolis.txt -s 2 -c 4
-------------------------------------------------------------
ipyrad [v.0.7.28]
Interactive assembly and analysis of RAD-seq data
-------------------------------------------------------------
loading Assembly: anolis
from saved path: ~/ipyrad-workshop/anolis.json
establishing parallel connection:
host compute node: [4 cores] on darwin
Step 2: Filtering reads
[####################] 100% processing reads | 0:01:02
The filtered files are written to a new directory called anolis_edits. Again,
you can look at the results output by this step and also some handy stats tracked
for this assembly.
## View the output of step 2
$ cat anolis_edits/s2_rawedit_stats.txt
reads_raw trim_adapter_bp_read1 trim_quality_bp_read1 reads_filtered_by_Ns reads_filtered_by_minlen reads_passed_filter
punc_IBSPCRIB0361 250000 108761 160210 66 12415 237519
punc_ICST764 250000 107320 178463 68 13117 236815
punc_JFT773 250000 110684 190803 46 9852 240102
punc_MTR05978 250000 102932 144773 54 12242 237704
punc_MTR17744 250000 103394 211363 55 9549 240396
punc_MTR21545 250000 119191 161709 63 21972 227965
punc_MTR34414 250000 109207 193401 54 16372 233574
punc_MTRX1468 250000 119746 134069 45 19052 230903
punc_MTRX1478 250000 116009 184189 53 16549 233398
punc_MUFAL9635 250000 114492 182877 61 18071 231868
## Get current stats including # raw reads and # reads after filtering.
$ ipyrad -p params-anolis.txt -r
You might also take a closer look at the filtered reads:
$ zcat anolis_edits/punc_IBSPCRIB0361.trimmed_R1_.fastq.gz | head -n 12
@D00656:123:C6P86ANXX:8:2201:3857:34366 1:Y:0:8
TGCATGTTTATTGTCTATGTAAAAGGAAAAGCCATGCTATCAGAGATTGGCCTGGGGGGGGGGGGCAAATACATG
+
;=11>111>1;EDGB1;=DG1=>1:EGG1>:>11?CE1<>1<1<E1>ED1111:00CC..86DG>....//8CDD
@D00656:123:C6P86ANXX:8:2201:5076:34300 1:N:0:8
TGCATATGAACCCCAACCTCCCCATCACATTCCACCATAGCAATCAGTTTCCTCTCTTCCTTCTTCTTGACCTCT
+
@;BFGEBCC11=/;/E/CFGGGG1ECCE:EFDFCGGGGGGG11EFGGGGGCGG:B0=F0=FF0=F:FG:FDG00:
@D00656:123:C6P86ANXX:8:2201:5042:34398 1:N:0:8
TGCATTCAAAGGGAGAAGAGTACAGAAACCAAGCACATATTTGAAAAATGCA
+
GGGGGGGCGGGGGGGGGGGGGEGGGFGGGGGGEGGGGGGGGGGGGGFGGGEG
This is actually really cool, because we can already see the results of both of our applied parameters. All reads have been trimmed to 75bp, and the third read had adapter contamination removed (you can tell because it’s shorter than 75bp). As an exercise you can go back up to the section where we looked at the raw data initially and see if you can identify the adapter sequence in this read. We will see more about the consequences of filtering adapters soon as well when we look at the clustered reads next.
Step 3: clustering within-samples
Step 3 de-replicates and then clusters reads within each sample by the
set clustering threshold and then writes the clusters to new files in a
directory called anolis_clust_0.85. Intuitively we are trying to
identify all the reads that map to the same locus within each sample.
The clustering threshold specifies the minimum percentage of sequence
similarity below which we will consider two reads to have come from
different loci.
The true name of this output directory will be dictated by the value you
set for the clust_threshold parameter in the params file.
You can see the default value is 0.85, so our default directory is named accordingly. This value dictates the percentage of sequence similarity that reads must have in order to be considered reads at the same locus. You’ll more than likely want to experiment with this value, but 0.85 is a reliable default, balancing over-splitting of loci vs over-lumping. Don’t mess with this until you feel comfortable with the overall workflow, and also until you’ve learned about Branching assemblies.
There have been many papers written comparing how results of assemblies vary depending on the clustering threshold. In general, my advice is to use a value between about .82 and .95. Within this region results typically do not vary too significantly, whereas above .95 you will oversplit loci and recover fewer SNPs.
It’s also possible to incorporate information from a reference genome to improve clustering at this step, if such a resources is available for your organism (or one that is relatively closely related). We will not cover reference based assemblies in this workshop, but you can refer to the ipyrad documentation for more information.
Note on performance: Steps 3 and 6 generally take considerably longer than any of the steps, due to the resource intensive clustering and alignment phases. These can take on the order of 10-100x as long as the next longest running step. This depends heavily on the number of samples in your dataset, the number of cores, the length(s) of your reads, and the “messiness” of your data in terms of the number of unique loci present (this can vary from a few thousand to many millions).
Now lets run step 3:
$ ipyrad -p params-anolis.txt -s 3 -c 2
-------------------------------------------------------------
ipyrad [v.0.7.28]
Interactive assembly and analysis of RAD-seq data
-------------------------------------------------------------
loading Assembly: anolis
from saved path: ~/ipyrad-workshop/anolis.json
establishing parallel connection:
host compute node: [2 cores] on darwin
Step 3: Clustering/Mapping reads
[####################] 100% dereplicating | 0:00:11
[####################] 100% clustering | 0:19:35
[####################] 100% building clusters | 0:00:06
[####################] 100% chunking | 0:00:01
[####################] 100% aligning | 0:14:27
[####################] 100% concatenating | 0:00:04```
In-depth operations of step 3:
- dereplicating - Merge all identical reads
- clustering - Find reads matching by sequence similarity threshold
- building clusters - Group similar reads into clusters
- chunking - Subsample cluster files to improve performance of alignment step
- aligning - Align all clusters
- concatenating - Gather chunked clusters into one full file of aligned clusters
Again we can examine the results. The stats output tells you how many clusters were found (‘clusters_total’), and the number of clusters that pass the mindepth thresholds (‘clusters_hidepth’). We’ll go into more detail about mindepth settings in some of the advanced tutorials but for now all you need to know is that by default step 3 will filter out clusters that only have a handful of reads on the assumption that these are probably all mostly due to sequencing error.
$ ipyrad -p params-anolis.txt -r
Summary stats of Assembly anolis
------------------------------------------------
state reads_raw reads_passed_filter clusters_total clusters_hidepth
punc_IBSPCRIB0361 3 250000 237519 56312 4223
punc_ICST764 3 250000 236815 60626 4302
punc_JFT773 3 250000 240102 61304 5214
punc_MTR05978 3 250000 237704 61615 4709
punc_MTR17744 3 250000 240396 62422 5170
punc_MTR21545 3 250000 227965 55845 3614
punc_MTR34414 3 250000 233574 61242 4278
punc_MTRX1468 3 250000 230903 54411 3988
punc_MTRX1478 3 250000 233398 57299 4155
punc_MUFAL9635 3 250000 231868 59249 3866
Again, the final output of step 3 is dereplicated, clustered files for
each sample in ./anolis_clust_0.85/. You can get a feel for what
this looks like by examining a portion of one of the files.
We’ll take a moment now to compare the outputs of the different empirical libraries.
## Same as above, `zcat` unzips and prints to the screen and
## `head -n 28` means just show me the first 28 lines.
$ zcat anolis_clust_0.85/punc_IBSPCRIB0361.clustS.gz | head -n 28
000e3bb624e3bd7e91b47238b7314dc6;size=4;*
TGCATATCACAAGAGAAGAAAGCCACTAATTAAGGGGAAAAGAAAAGCCTCTGATATAGCTCCGATATATCATGC-
75e462e101383cca3db0c02fca80b37a;size=2;-
-GCATATCACAAGAGAAGAAAGCCACTAATTAAGGGGAAAAGAAAAGCCTCTGATATAGCTCCGATATATCATGCA
//
//
0011c57e1e3c03e4a71516bd51c623da;size=1;*
TGCATGAAATAGATACAACTGAGCACATTTGCTTTGTTTCCAGAGAGTGCAACAAGAGTTTGGAGAATATAAATG
eef50f7e4849ed4761f1fd38b08d0e12;size=1;+
TGCATGAAATAGATACTACTGAGCACATTTGCTTTGTTTCCAGAGATTGCATCAAGAGTTTGGAGAATATAAATG
7f089b34522da8288b0e6ff7db8ffc6c;size=1;+
TGCATGAAATAGATACAACTGAGCACATTTGCTTTGTTTCCAGAGATTGCAACAAGAGTTTGGAGAATATAAATG
//
//
001236a2310c39a3a16d96c4c6c48df1;size=4;*
TGCATCTCTTTGGGCTGTTGCTTGGTGGCACACCATGCTGCTTTCTCCTCACTTTTTCTCTCTTTTCCTGAGACT------------------------------
4644056dca0546a270ba897b018624b4;size=2;-
------------------------------CACCATGCTGCTTTCTCCTCACTTTTTCTCTCTTTTCCTGAGACTGAGCCAGGGACAGCGGCTGAGGAGGATGCA
5412b772ec0429af178caf6040d2af30;size=1;+
TGCATTTCTTTGGGCTGTTGCTTGGTGGCACACCATGCTGCTTTCTCCTCACTTTTTCTCTCTTTTCCTGAGACT------------------------------
//
//
0013684f0db0bd454a0a6fd1b160266f;size=1;*
TGCATTGTTCATGAATCGTCCCATTGTATACATTTTACCTGATCTATCTCATTGTATTTTACTCCATGGTTTTCA-------------------------
c26ec07b3e3e77d3167341d100fd2d4e;size=1;-
-------------------------GTATACATTTTACTTGATCTATCTCATTGTATTTTACTCCATGGTTTTCAGTACCTAACAAGCAGCATGTATGCA
55510205b75b441a2c3ce6249f1eb47c;size=1;-
-------------------------GTATACATTTTACCTGATCTATCTTATTGTATTTTACTCCATGGTTTTCAGTACCTAACAAGCAGCATGTATGCA
Reads that are sufficiently similar (based on the above sequence similarity threshold) are grouped together in clusters separated by “//”. For the second and fourth clusters above these are probably homozygous with some sequencing error, but it’s hard to tell. For the first and third clusters, are there truly two alleles (heterozygote)? Is it a homozygote with lots of sequencing errors, or a heterozygote with few reads for one of the alleles?
Thankfully, untangling this mess is what step 4 is all about.