FastQC for quality control
To get a better view of the data quality, without looking at individual reads by hand like we did earlier, we use automated approaches to check the quality. We will use FastQC to generate a sample-wide summary of data quality.
The logic of FastQC is that we want to obtain a high-level view of the quality of the sequencing. You may be able to detect low quality samples, but if you have a lot of samples, you may not want to run FastQC for every single file. Even running it for a few samples will give you good insight into overall quality of the sequencing run. For example, a key QC procedure involves inspecting average quality scores per base position and trimming read edges, which is where low quality base-calls tend to accumulate. In this figure, the X-axis shows the position on the read in base-pairs and the Y-axis depicts information about Phred quality score per base for all reads, including median (center red line), IQR (yellow box), and 10%-90% (whiskers). As an example, here is a very clean base sequence quality report for a 75bp RAD-Seq library. These reads have generally high quality across their entire length, with only a slight (barely worth mentioning) dip toward the end of the reads:
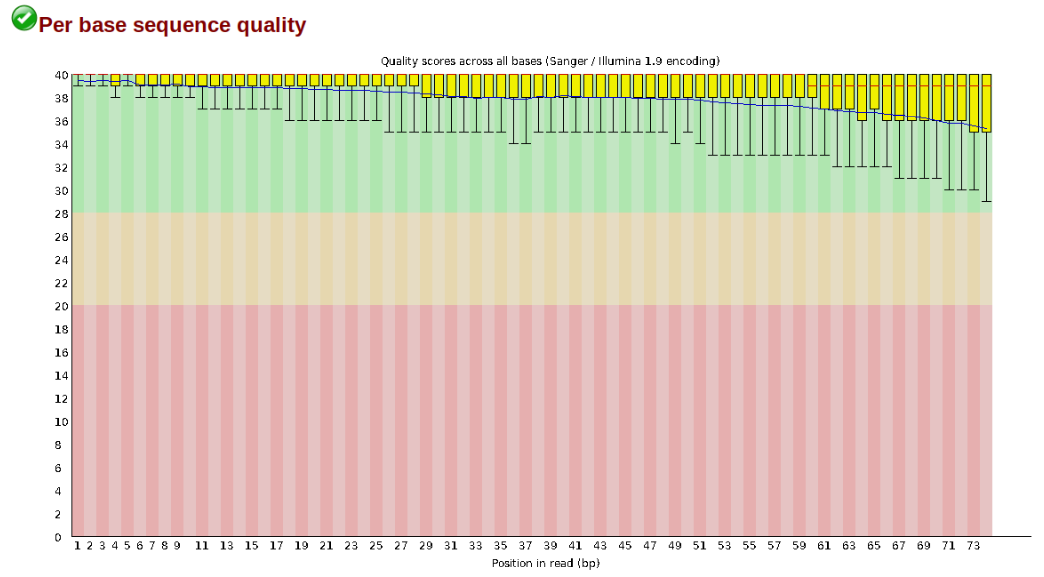
In contrast, here is a somewhat typical base sequence quality report for R1 of a 300bp paired-end Illumina run of ezRAD data:
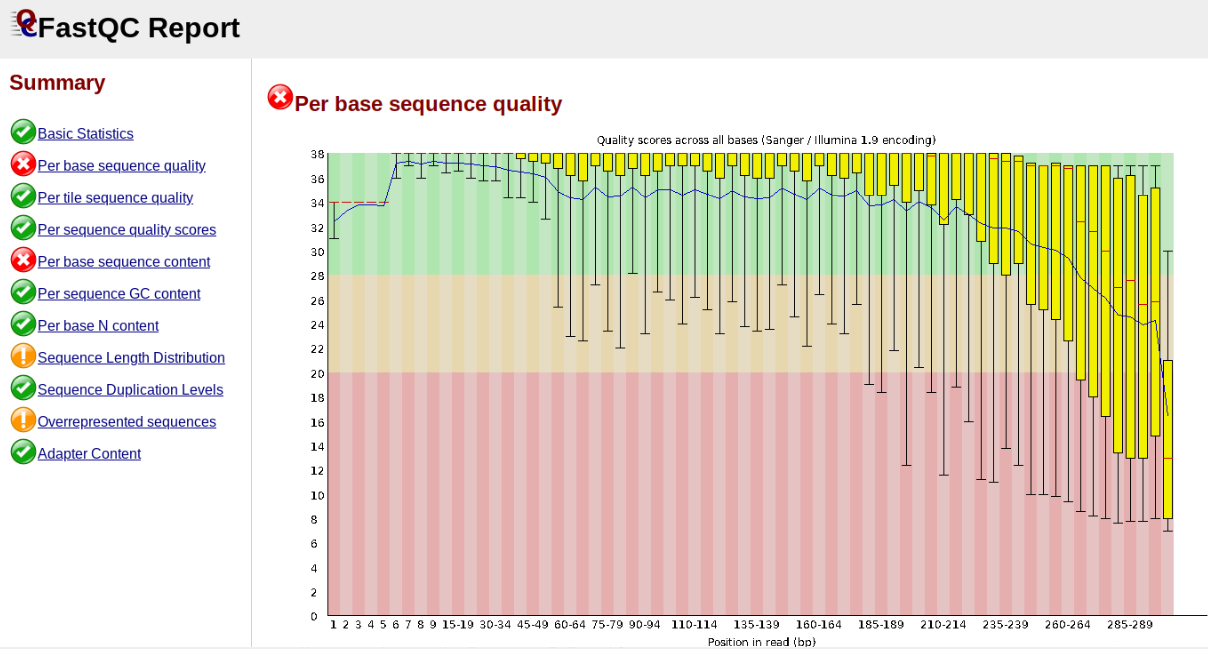
This figure depicts a common artifact of current Illumina chemistry, whereby quality scores per base drop off precipitously toward the ends of reads, with the effect being magnified for read lengths > 150bp. The purpose of using FastQC to examine reads is to determine whether and how much to trim our reads to reduce sequencing error interfering with basecalling. In the above figure, as in most real dataset, we can see there is a tradeoff between throwing out data to increase overall quality by trimming for shorter length, and retaining data to increase value obtained from sequencing with the result of increasing noise toward the ends of reads.
NB: Part of the point of running fastqc is to determine where the error
rate is too high in the sequences to reliably call bases. Once this is determined
one would then use the trim_reads parameter in ipyrad to remove these low quality
regions. In the above example we might choose to trim these reads to ~200bp.
First, install FastQC:
$ conda install -c bioconda fastqc -y
The ‘-y’ flag tells conda “Yes, please just do this and don’t ask me any questions.” The ‘-c bioconda’ flag tells conda to search for fastqc in the bioconda channel. Conda has many many channels, which are repositories of different packages.
Now run FastQC on this sample:
$ cd /scratch/ipyrad-workshop
$ fastqc Amaranthus_R1_.fastq.gz
FastQC will indicate its progress in the terminal. This toy data will run quite quickly, but real data can take somewhat longer to analyse (10s of minutes).
Started analysis of Amaranthus_R1_.fastq.gz
Approx 5% complete for Amaranthus_R1_.fastq.gz
Approx 10% complete for Amaranthus_R1_.fastq.gz
Approx 15% complete for Amaranthus_R1_.fastq.gz
Approx 20% complete for Amaranthus_R1_.fastq.gz
Approx 25% complete for Amaranthus_R1_.fastq.gz
Approx 30% complete for Amaranthus_R1_.fastq.gz
Approx 35% complete for Amaranthus_R1_.fastq.gz
Approx 40% complete for Amaranthus_R1_.fastq.gz
Approx 45% complete for Amaranthus_R1_.fastq.gz
Approx 50% complete for Amaranthus_R1_.fastq.gz
Approx 55% complete for Amaranthus_R1_.fastq.gz
Approx 60% complete for Amaranthus_R1_.fastq.gz
Approx 65% complete for Amaranthus_R1_.fastq.gz
Approx 70% complete for Amaranthus_R1_.fastq.gz
Approx 75% complete for Amaranthus_R1_.fastq.gz
Approx 80% complete for Amaranthus_R1_.fastq.gz
Approx 85% complete for Amaranthus_R1_.fastq.gz
Approx 90% complete for Amaranthus_R1_.fastq.gz
Approx 95% complete for Amaranthus_R1_.fastq.gz
Approx 100% complete for Amaranthus_R1_.fastq.gz
Analysis complete for Amaranthus_R1_.fastq.gz
FastQC will save the output as an html file in the folder you’re currently in.
You want to look at it in your browser window. So, go back to the jupyter dashboard
and navigate to /home/ipyrad-workshop/ and click on Amaranthus_R1__fastqc.html.
This will open the FastQC report which provides extensive information about
the quality of the data, which we will briefly review here.
Inspecting and Interpreting FastQC Output
Opening up this html file, on the left you’ll see a summary of all the results, which highlights areas FastQC indicates may be worth further examination. We will only look at a few of these.

Lets start with Per base sequence quality.
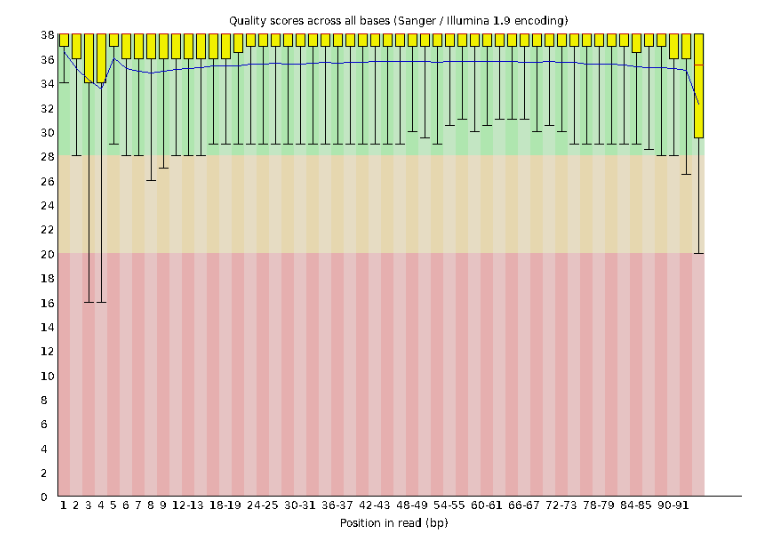
For this data the sequence quality per base is uniformly quite high, with dips only in the first and last 5 bases (again, this is typical for Illumina reads). Based on information from this plot we can see that this data doesn’t need any trimming, which is good.
Now lets look at the Per base sequece content, which FastQC highlights with a
scary red X.
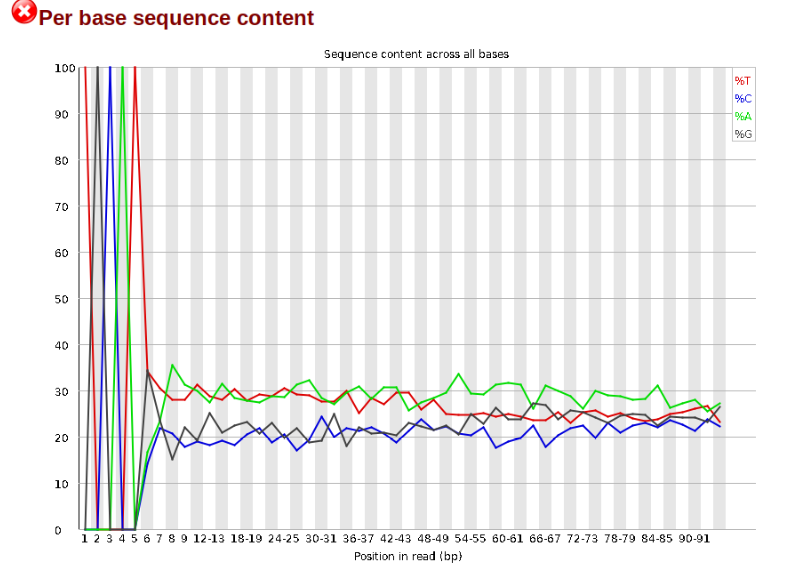
The squiggles indicate base composition per base position averaged across the
reads. It looks like the signal FastQC is concerned about here is related to
the extreme base composition bias of the first 5 positions. We happen to know
this is a result of the restriction enzyme overhang present in all reads
(TGCAT in this case for the EcoT22I enzyme used), and so it is in fact of no
concern. Now lets look at Adapter Content:
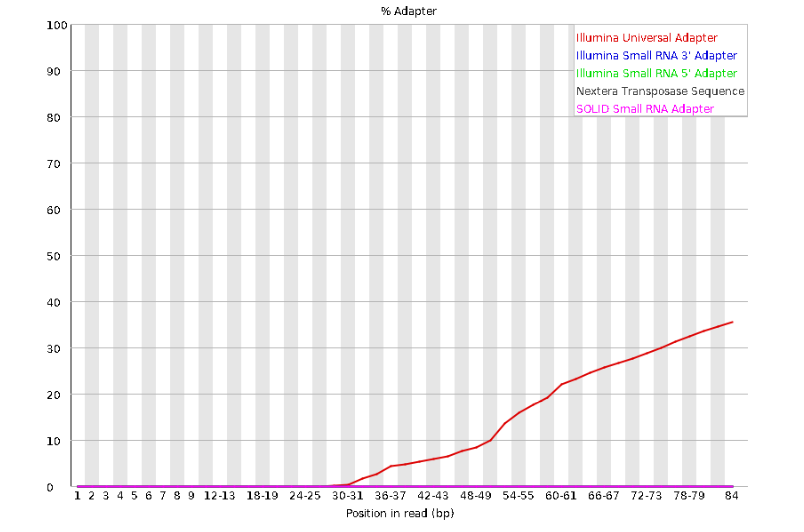
Here, we can see adapter contamination increases toward the tail of the reads, approaching 40% of total read content at the very end. The concern here is that if adapters represent some significant fraction of the read pool, then they will be treated as “real” data, and potentially bias downstream analysis. In the Amaranthus data this looks like it might be a real concern so we shall keep this in mind during step 2 of the ipyrad analysis, and incorporate 3’ read trimming and aggressive adapter filtering.
Running fastqc on empirical data
Now we will run fastqc on a couple of samples from the empirical data. To do
this we will first change directory (cd) to where the demultiplexed samples
live, inside /data/Step1-Results.
$ cd /data/Step1-Results/Alaasam/Alaasam_fastqs
$ fastqc 07089_C18_5_R1_.fastq.gz -o /scratch/ipyrad-workshop/
$ fastqc 07089_C18_5_R1_.fastq.gz -o /scratch/ipyrad-workshop/
NB: The data assets attached to capsules in CO are ‘read-only’ meaning
we have to write the fastqc outputs to some other directory. This is what the
-o flag is telling fastqc, to write the outputs to /scratch/ipyrad-workshop.
In the remaining time, consult with your group members and each choose 1-2 samples to run fastqc on both R1 and R2. After the runs finish, inspect the results and try to come to a consensus about what the results indicate.
Be prepared to answer the following questions:
- Were there any significant quality issues with any of the samples?
- Will you choose to use
trim_readsto remove low quality regions? If so what values? - Was there noticeable adapter contamination?