Demographic inference using the Site Frequency Spectrum (SFS) with momi2
What is the SFS?
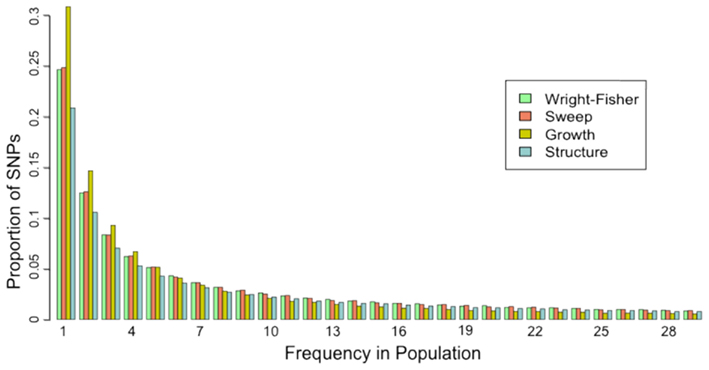
The site frequency spectrum (SFS) is a histogram of the frequencies of SNPs in a sample of individuals from a population. Different population histories leave characteristic signatures on the SFS. For example, a population that has undergone a recent bottleneck will have a reduced number of rare variants as compared to a neutrally evolving population. Rare variants will be lost much more rapidly than common variants under a bottleneck model. On the other hand, population expansion models will display an excess of rare variants with respect to a neutral model. In a similar fashion, selection and gene flow can leave characteristic imprints on the SFS of a population. A good example of how the SFS is calculated can be found on wikipedia.
What is demographic inference?
The goal of demographic analyses is to understand the history of lineages (sometimes referred as ‘populations’) in a given species, estimating the neutral population dynamics such as time of divergence, population expansion, population contraction, bottlenecks, admixture, etc. A nice example of a paper that performs model selection and parameter estimation Portik et al 2017.
Most importantly, how do you pronounce momi?
Pronunciation: Care of Jonathan Terhorst (somewhat cryptically), from a github issue I created to resolve this conundrum: “How do you pronounce ∂a∂i? ;-)”…. And another perspective from Jack Kamm: “Both pronunciations are valid, but I personally say ‘mommy’”.
Prerequisites
This notebook code assumes you already have a jupyter notebook running in python3.
momi2 installation
momi2 requires python3, which is a different version of python we’ve been using up to now. Fortunately conda makes it easy to run python2 and python3 side by side. We will install python3 in a separate conda environment, and then install and run momi2 analyses using this environment. A conda environment is a container for python packages and configurations. More on creating/managing conda environments can be found here.
Begin by opening an ssh session on the cluster and creating our new environment:
## -n assigns a name to the environment
## python=3.6 specifies the python version of the new environment
$ conda create -n momi-py36 python=3.6
After the install finishes you can inspect the currently available environments:
$ conda env list
# conda environments:
#
base * /home/isaac/miniconda2
momi-py36 /home/isaac/miniconda2/envs/momi-py36
And now switch to the new python3 environment:
$ source activate momi-py36
(momi-py36) [work2@watson:~]$
Note: You’ll notice that the conda env you are currently using is now displayed as part of your prompt. We will maintain this convention for the rest of this notebook.
Now use conda to install momi2 and jupyter. All the -c arguments again are specifying
channels that momi2 pulls dependencies from. Order matters here, so copy and paste this
command to your terminal.
(momi-py36)$ conda install momi ipyparallel openblas jupyter -c defaults -c conda-forge -c bioconda -c jackkamm -y
This will produce copious output, and should take ~5-10 minutes.
momi2 Analyses
Create a new notebook inside your ~/ipyrad-workshop/ directory
called anolis-momi2.ipynb (refer to the jupyter notebook configuration page for a refresher on connecting to the notebook server). The rest of the
materials in this part of the workshop assume you are running all code
in cells of a jupyter notebook that is running on the cluster.
- Constructing and plotting a simple model
- Preparing real data for analysis
- Specific considerations when using RAD-like data with momi2
- Inference procedure
- Bootstrapping confidence intervals
Constructing and plotting a simple model
One of the real strengths of momi2 is the ability not only to construct a demographic history for a set of populations, but also to plot the model to verify that it corresponds to what you expect!
Begin with the usual import statements, except this time we also add logging,
which allows momi2 to write progress to a log file. This can be useful for
debugging, so we encourage this practice.
%matplotlib inline
import momi ## momi2 analysis
import logging ## create log file
logging.basicConfig(level=logging.INFO,
filename="momi_log.txt")
A demographic model is composed of leaf nodes, migration events,
and size change events. We start with the simplest possible 2
population model, with no migration, and no size changes. For the
sake of demonstrating model construction we choose arbitrary
values for N_e (the diploid effective size), and t (the time
at which all lineages move from the “South” population to the
“North” population).
model = momi.DemographicModel(N_e=1e5)
model.add_leaf("North")
model.add_leaf("South")
model.move_lineages("South", "North", t=2e5)
Note: The default migration fraction of the
DemographicModel.move_lineages()function is 100%, so if we do not specify this value then when we callmove_lineagesmomi2 assumes we want to move all lineages from the source to the destination. Later we will see how to manipulate the migration fraction to only move some portion of lineages.
Executing this cell produces no output, but that’s okay, we are just specifying the model. Also, be aware that the names assigned to leaf nodes have no specific meaning to momi2, so these names should be selected to have specific meaning to your target system. Here “North” and “South” are simply stand-ins for some hypothetical populations. Now that we have this simple demographic model parameterized we can plot it, to see how it looks.
yticks = [1e4, 2.5e4, 5e4, 7.5e4, 1e5, 2.5e5, 5e5, 7.5e5]
fig = momi.DemographyPlot(
model,
["North", "South"],
figsize=(6,8),
major_yticks=yticks,
linthreshy=1e5)
There’s a little bit going on here, but we’ll walk you through it:
yticks- This is a list of elements specifying the timepoints to highlight on the y-axis of the figure.
The first two arguments to momi.DemographyPlot() are required, namely the model to plot, and the populations of the model to include. The next three arguments are optional, but useful:
figsize- Specify the output figure size as (width, height) in inches.major_yticks- Tells the momi2 plotting routine to use the time demarcations we specified in thieyticksvariable.linthreshy- The time point at which to switch from linear to log-scale, backwards in time. This is really useful if you have many “interesting” events happening relatively recently, and you don’t want them to get “smooshed” together by the depth of the older events. This will become clearer as we add migration events later in the tutorial.
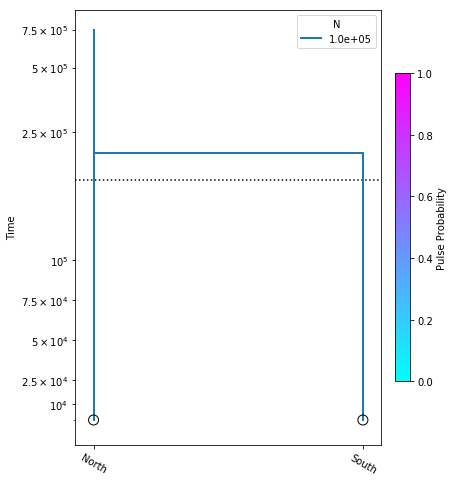
Experiment: Try changing the value of linthreshy and replotting. Try 1e4 and 1.5e5 and notice how the figure changes. You can also experiment with changing the values in the yticks list.
Let’s create a new model and introduce one migration event that only moves some fraction of lineages, and not the totality of them:
model = momi.DemographicModel(N_e=1e5)
model.add_leaf("North")
model.add_leaf("South")
model.move_lineages("South", "North", p=0.1, t=5e4)
model.move_lineages("South", "North", t=2e5)
yticks = [1e4, 2.5e4, 5e4, 7.5e4, 1e5, 2.5e5, 5e5, 7.5e5]
fig = momi.DemographyPlot(
model, ["North", "South"],
figsize=(6,8),
major_yticks=yticks,
linthreshy=1.5e5)
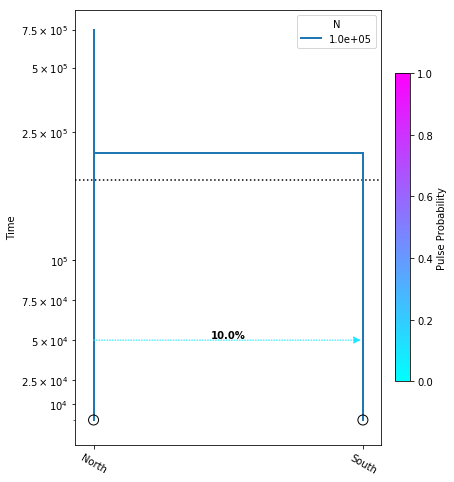
This is almost the exact same model as above, except now we have introduced another move_lineages call which includes the p=0.1 argument. This indicates that we wish to move 10% of lineages from the “South” population to the “North” population at the specified timepoint.
Note: It may seem odd that the arrow in this figure points from “North” to “South”, but this is simply because we are operating in a coalescent framework and therefore the
move_lineagesfunction operates backwards in time.
Preparing real data for analysis
We need to gather and construct several input files before we can actually apply momi2 to our Anolis data.
- Population assignment file - This is a tab or space separated list of sample names and population names to which they are assigned. Sample names need to be exactly the same as they are in the VCF file. Population names can be anything, but it’s useful if they’re meaningful.
- Properly formatted VCF - We do have the VCF file output from the ipyrad Anolis assembly, but it requires a bit of massaging before it’s ready for momi2. It must be zipped and indexed in such a way as to make it searchable.
- BED file - This file specifies genomic regions to include in when calculating the SFS. It is composed of 3 columns which specify ‘chrom’, ‘chromStart’, and ‘chromEnd’.
- The allele counts file - The allele counts file is an intermediate file that we must generate on the way to constructing the SFS. momi2 provides a function for this.
- Genereate the SFS - The culmination of all this housekeeping is the SFS file which we will use for demographic inference.
Population assignment file
Based on the results of the PCA and also our knowledge of the geographic location of the samples we will assign 2 samples to the “North” population, and 8 samples to the “South” population. To save some time we created this pops file, and have stashed a copy in the /scratch/af-biota directory. We can simply copy the file from there into our own ipyrad-workshop directories. We could do this by finding a terminal on the cluster, but its also possible to run terminal commands from jupyter notebooks using “magic” commands. Including %%bash on the first line of a cell tell jupyter to interpret lines inside this cell as terminal commands, so we can do this:
%%bash
cp /rigel/edu/radcamp/users/work1/ipyrad-workshop/anolis_pops.txt .
cat anolis_pops.txt
punc_ICST764 North
punc_MUFAL9635 North
punc_IBSPCRIB0361 South
punc_JFT773 South
punc_MTR05978 South
punc_MTR17744 South
punc_MTR21545 South
punc_MTR34414 South
punc_MTRX1468 South
punc_MTRX1478 South
Magic!
Note:
catis a command line utility that prints the contents of a file to the screen.
Properly formatted VCF
In this tutorial we are using a very small dataset, so manipulating the VCF is very fast. With real data the VCF file can be enormous, which makes processing it very slow. momi2 expects very large input files, so it insists on having them preprocessed to speed things up. The details of this preprocessing step are not very interesting, but we are basically compressing and indexing the VCF so it’s faster to search.
%%bash
## bgzip performs a blockwise compression
## The -c flag directs bgzip to leave the original vcf file
## untouched and create a new file for the vcf.gz
cp /rigel/edu/radcamp/users/work1/ipyrad-workshop/anoles_outfiles/anoles.vcf anolis.vcf
bgzip -c anolis.vcf > anolis.vcf.gz
## tabix indexes the file for searching
tabix anolis.vcf.gz
ls
anolis.alleles.loci anolis.loci anolis.snps.phy anolis.u.snps.phy
anolis.geno anolis.nex anolis_stats.txt anolis.ustr
anolis.gphocs anolis.phy anolis.str anolis.vcf.gz
anolis.hdf5 anolis.snps.map anolis.u.geno anolis.vcf.gz.tbi
BED file
The last file we need to construct is a BED file specifying which genomic regions to retain for calculation of the SFS. The standard coalescent assumes no recombination and no natural selection, so drift and mutation are the only forces impacting allele frequencies in populations. If we had whole genome data, and a good reference sequence then we would have information about coding regions and other things that are probably under selection, so we could use the BED file to exclude these regions from the analysis. With RAD-Seq type data it’s very common to assume RAD loci are neutrally evolving and unlinked, so we just want to create a BED file that specifies to retain all our SNPs. We provide a simple python program to do this conversion, which is located in the /scratc/af-biota/bin directory.
%%bash
/rigel/edu/radcamp/users/work1/bin/vcf2bed.py anolis.vcf anolis.bed
## Print the first 10 lines of this file
head anoles.bed
locus_1 7 8
locus_3 65 66
locus_5 13 14
locus_5 26 27
locus_5 55 56
locus_8 34 35
locus_21 13 14
locus_24 58 59
locus_26 2 3
locus_26 50 51
The allele counts file
The allele counts file is an intermediate step necessary for generating the SFS. It’s a format internal to momi2, so we won’t spend a lot of time describing it, except to say that it is exactly what it says it is: A count of alleles in each population. Since each diploid individual has 2 alleles per snp, the total count of alleles per population will be 2n at maximum, and 0 at minimum.
%%bash
python -m momi.read_vcf --no_aa --verbose anolis.vcf.gz anolis_pops.txt anolis_allele_counts.gz --bed anolis.bed
gunzip -c anolis_allele_counts.gz | head
/home/isaac/miniconda2/envs/momi-py36/bin/python
{
"populations": ["North", "South"],
"use_folded_sfs": true,
"length": 1911,
"n_read_snps": 148,
"n_excluded_snps": 0,
"configs": [
[[0, 0], [1, 1]],
[[0, 0], [3, 1]],
[[0, 0], [2, 2]],
Generate the SFS
The momi site frequency spectrum is represented somewhat differently than you might be used to if you have used dadi or fastsimcoal2. Here we load the SFS generated above into the sfs object and print a few properties.
%%bash
python -m momi.extract_sfs anolis_sfs.gz 20 anolis_allele_counts.gz
sfs = momi.Sfs.load("anolis_sfs.gz")
print("Avg pairwise heterozygosity", sfs.avg_pairwise_hets[:5])
print("populations", sfs.populations)
print("percent missing data per population", sfs.p_missing)
Using RAD data with momi
Thanks to a fruitful tele-visit by Jack Kamm to the AMNH ECEM meeting on 10/3/18, here are some momi tips for the non-model RAD-seq crowd:
- Missing data: Momi correctly handles missing data, but the parameter estimates it produces are scaled by the average number of pairwise differences between samples, rather than by the total tree length, so care should be taken when interpreting them. If you don’t know the mutation rate or the generation time there’s a scaling invariance. Relative times are all equivalent given a constant scaling factor.
- “Real” time: If you want parameter estimates in “real” time you must set
muts_per_genandgen_timeon your Demographic Model, and make sure you accurately set thelengthparameter and setuse_pairwise_diffs=Falsewhen you call DemographicModel.set_data(). If you have missing data you must remove this by using theSnpAlleleCounts.down_sample()function, which is analogous to the projection method in dadi. - Ensure correct ‘length’: The length of the SFS is calculated from the bed file during the SnpAlleleCounts.read_vcf() call. If your bed file only records snps, then the true sequence length is not reflected and you should be sure to set the
lengthargument when you call model.set_data(). This value should equal the total number of basepairs of your concatenated RAD-seq dataset, i.e. nloci * locus_length if all loci are equal length. - Sample sizes: There is some doubt in the manuscript about the max number of samples per population. It seems that on the order of tens of samples per population is fine. Around 50 seems fine, but more than 100 and it starts to get dicey.
- Continuous migration: Adding multiple migration pulses improves the fit of momi models to data generated under a continuous migration scenario.
- Divergence depth: Works okay for human/neandertal scale divergence but not so much for human/chimp.
- Time: Inferring timing of events is generally a difficult thing. Migration pulse times are the least most difficult of all the parameters to estimate.
- More populations: Adding additional populations can add a lot of additional power. You can see things that happened more distantly in the past when those populations merged together.
- Migration event penalty: Computation time scales linearly with number of migration events, which is good, but does have a memory cost.
-
Optimization strategy: In general L-BFGS-B is preferred over the default TNC. TNC will always work tenaciously to get the best result, but L-BFGS-B will get you a good enough result much faster.
- Validation: Because the SFS is a summary of the data, conditions exist where the SFS may non-identifiable, or you may find degenerate/runaway behavior. It is always important to validate your inference with simulations and bootstrapping.
Inference procedure
In the previous examples where we constructed and plotted DemographicModels, we had specified all the values for population sizes, divergence times, and migration fractions. This is useful when we are developing the models we want to test, because we can construct the model with toy parameter values, plot it and then visually inspect whether the model meets our expectations. Once we have settled on one or a handful of models to test, we can incorporate the observed SFS in an inference procedure in order to test which model is the best fit to the data. The best fitting model will then provide a set of maximum likelihood parameter values for the parameters we are interested in (like divergence time). We can then perform a bootstrap analysis, by randomly resampling the observed SFS, re-estimating parameters under the most likely model, and constructing bootstrap confidence intervals on these values (typically 50-100 replicates, but here 10 for speed).
Here we will invesigate three different 2 population models:
no_migration_model- All parameters fixed, except divergence time.pop_sizes_model- North and South populations are allowed to have different, variable sizes. Here we also estimate divergence time.migration_model- Allow one pulse of migration in both directions, at possibly different times, and with different migration fractions. Also, include all other parameters above (population sizes and divergence time).
Estimating divergence time
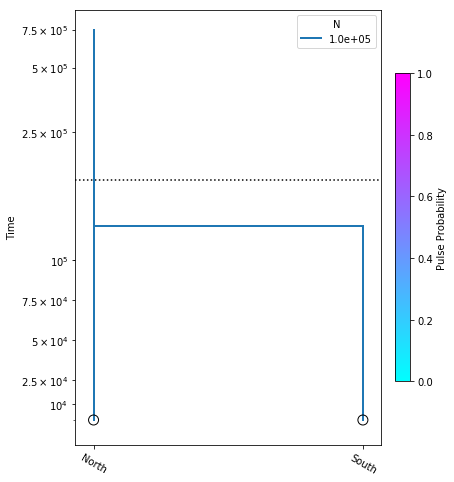
Here we construct the no_migration_model, where we are estimating only divergence time. We perform the optimization, and plot the model with the resulting most likely parameter value.
no_migration_model = momi.DemographicModel(N_e=1e5)
no_migration_model.set_data(sfs)
no_migration_model.add_time_param("tdiv")
no_migration_model.add_leaf("North")
no_migration_model.add_leaf("South")
no_migration_model.move_lineages("South", "North", t="tdiv")
no_migration_model.optimize()
fun: 0.6331043898321572
jac: array([-2.03580439e-13])
kl_divergence: 0.6331043898321572
log_likelihood: -252.5589124720131
message: 'Converged (|f_n-f_(n-1)| ~= 0)'
nfev: 11
nit: 3
parameters: ParamsDict({'tdiv': 121612.07225824424})
status: 1
success: True
x: array([121612.07225824])
yticks = [1e4, 2.5e4, 5e4, 7.5e4, 1e5, 2.5e5, 5e5, 7.5e5]
fig = momi.DemographyPlot(
no_migration_model, ["North", "South"],
figsize=(6,8),
major_yticks=yticks,
linthreshy=1.5e5)
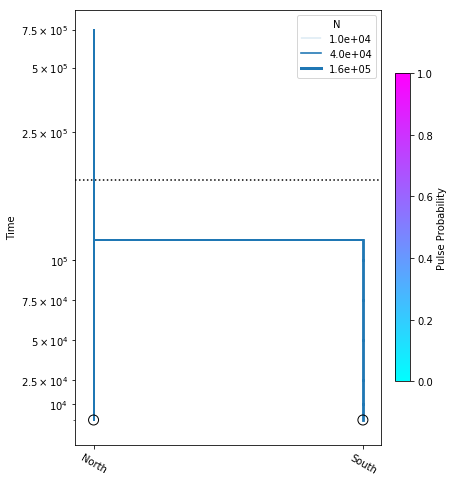
Including population size parameters
Here we construct the popsizes_model, where we are estimating variable population sizes as well as divergence time. We perform the optimization, and plot the model with the resulting most likely parameter values.
popsizes_model = momi.DemographicModel(N_e=1e5)
popsizes_model.set_data(sfs)
popsizes_model.add_size_param("n_north")
popsizes_model.add_size_param("n_south")
popsizes_model.add_time_param("tdiv")
popsizes_model.add_leaf("North", N="n_north")
popsizes_model.add_leaf("South", N="n_south")
popsizes_model.move_lineages("South", "North", t="tdiv")
popsizes_model.optimize()
fun: 0.6080302639574219
jac: array([-7.11116810e-06, 1.25870299e-06, 5.18524040e-11])
kl_divergence: 0.6080302639574219
log_likelihood: -248.84794184255227
message: 'Converged (|f_n-f_(n-1)| ~= 0)'
nfev: 13
nit: 6
parameters: ParamsDict({'n_north': 58437.32067443618, 'n_south': 132874.21082953943, 'tdiv': 112867.76818828644})
status: 1
success: True
x: array([1.09757100e+01, 1.17971582e+01, 1.12867768e+05])
yticks = [1e4, 2.5e4, 5e4, 7.5e4, 1e5, 2.5e5, 5e5, 7.5e5]
fig = momi.DemographyPlot(
popsizes_model, ["North", "South"],
figsize=(6,8),
major_yticks=yticks,
linthreshy=1.5e5)
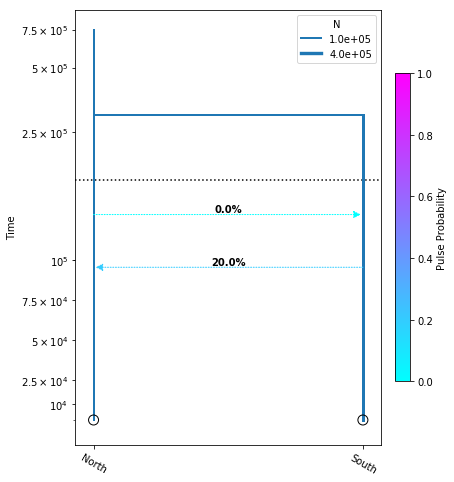
Adding migration events
migration_model = momi.DemographicModel(N_e=1e5)
migration_model.set_data(sfs)
migration_model.add_time_param("tmig_north_south")
migration_model.add_time_param("tmig_south_north")
migration_model.add_pulse_param("mfrac_north_south", upper=.2)
migration_model.add_pulse_param("mfrac_south_north", upper=.2)
migration_model.add_size_param("n_north")
migration_model.add_size_param("n_south")
migration_model.add_time_param("tdiv", lower_constraints=["tmig_north_south", "tmig_south_north"])
migration_model.add_leaf("North", N="n_north")
migration_model.add_leaf("South", N="n_south")
migration_model.move_lineages("North", "South", t="tmig_north_south", p="mfrac_north_south")
migration_model.move_lineages("South", "North", t="tmig_south_north", p="mfrac_south_north")
migration_model.move_lineages("South", "North", t="tdiv")
migration_model.optimize()
fun: 0.6072433390803555
jac: array([-4.44751606e-11, 1.40562352e-11, -2.46501280e-04, 1.24640859e-08,
-2.14858289e-08, 2.17343690e-12, 1.43063167e-11])
kl_divergence: 0.6072433390803555
log_likelihood: -248.73147696074645
message: 'Converged (|f_n-f_(n-1)| ~= 0)'
nfev: 31
nit: 8
parameters: ParamsDict({'tmig_north_south': 95596.04975739817, 'tmig_south_north': 128584.2961865477, 'mfrac_north_south': 0.2, 'mfrac_south_north': 5.497639676362986e-07, 'n_north': 101409.03648320214, 'n_south': 274995.4822711156, 'tdiv': 300936.29078211787})
status: 1
success: True
x: array([ 9.55960498e+04, 1.28584296e+05, -1.38629436e+00, -1.44137763e+01,
1.15269175e+01, 1.25245099e+01, 1.72351995e+05])
yticks = [1e4, 2.5e4, 5e4, 7.5e4, 1e5, 2.5e5, 5e5, 7.5e5]
fig = momi.DemographyPlot(
migration_model, ["North", "South"],
figsize=(6,8),
major_yticks=yticks,
linthreshy=1.5e5)
Model selection with AIC
Model selection is typically performed with AIC, so here we extract the log likelihood of each model, calculate the AIC, and then calculate delta AIC values, and AIC weights. The best model will have the lowest AIC score. Delta AIC, and the AIC weight are indications of how confident we can be that the best fitting model is the correct model.
import numpy as np
AICs = []
for model in [no_migration_model, popsizes_model, migration_model]:
lik = model.log_likelihood()
nparams = len(model.get_params())
aic = 2*nparams - 2*lik
print("AIC {}".format(aic))
AICs.append(aic)
minv = np.min(AICs)
delta_aic = np.array(AICs) - minv
print("Delta AIC per model: ", delta_aic)
print("AIC weight per model: ", np.exp(-0.5 * delta_aic))
AIC 3857.113601374685
AIC 3859.6581687120392
AIC 3852.411537337999
[4.70206404 7.24663137 0. ]
[0.09527079 0.02669402 1. ]
Bootstrapping confidence intervals
We will use a bootstrap procedure to construct confidence intervals on parameters from our best model. Here we will run 10 bootstraps, for the sake of time, but on real data you would normally perform 50-100 bootstraps.
n_bootstraps = 10
# make copies of the original model to avoid changing them
no_migration_copy = no_migration_model.copy()
bootstrap_results = []
for i in range(n_bootstraps):
print(f"Fitting {i+1}-th bootstrap out of {n_bootstraps}")
# resample the data
resampled_sfs = sfs.resample()
# tell models to use the new dataset
no_migration_copy.set_data(resampled_sfs)
#add_pulse_copy.set_data(resampled_sfs)
# choose new random parameters for submodel, optimize
no_migration_copy.set_params(randomize=True)
no_migration_copy.optimize()
# initialize parameters from submodel, randomizing the new parameters
#add_pulse_copy.set_params(pulse_copy.get_params(),
#randomize=True)
#add_pulse_copy.optimize()
bootstrap_results.append(no_migration_copy.get_params())
Fitting 1-th bootstrap out of 10
Fitting 2-th bootstrap out of 10
Fitting 3-th bootstrap out of 10
Fitting 4-th bootstrap out of 10
Fitting 5-th bootstrap out of 10
Fitting 6-th bootstrap out of 10
Fitting 7-th bootstrap out of 10
Fitting 8-th bootstrap out of 10
Fitting 9-th bootstrap out of 10
Fitting 10-th bootstrap out of 10
fig = momi.DemographyPlot(
no_migration_model, ["North", "South"],
linthreshy=1e5, figsize=(6,8),
major_yticks=yticks,
draw=False)
# plot bootstraps onto the canvas in transparency
for params in bootstrap_results:
fig.add_bootstrap(
params,
# alpha=0: totally transparent. alpha=1: totally opaque
alpha=1/10)
# now draw the inferred demography on top of the bootstraps
fig.draw()
fig.draw_N_legend(loc="upper right")

In this figure the thick blue lines indicate the maximum likelihood values estimated under the best model, and the faint lines illustrate results of each of the ten bootstraps.
radcamp 2018 AF-Biota Feedback link
Please help us improve future radcamp workshops by completing this anonymous survey:
References
Portik, D. M., Leaché, A. D., Rivera, D., Barej, M. F., Burger, M., Hirschfeld, M., … & Fujita, M. K. (2017). Evaluating mechanisms of diversification in a Guineo‐Congolian tropical forest frog using demographic model selection. Molecular ecology, 26(19), 5245-5263.
Acknowledgements
We relied heavily on the excellent momi2 documentation during the creation of this tutorial.